Kistik fibrozis, son zamanlarda oldukça sık duymaya başladığımız bir hastalık. Gerek tanınmış insanların gerek küçük çocukların etkilenmesinden dolayı üzerindeki merak da arttı. Kistik fibrozis(CF), otozomal resesif bir kalıtsal hastalık olup beyaz ırkta en çok karşılaşılan genetik bir hastalıktır. 7. kromozomun üstünde bulunan Cystic Fibrozis Transmembrane Conductance Regulator (kistik fibrozis transmembran iletim regülatörü) adlı genin çeşitli mutasyonlara uğraması sonucu görülür. Günümüzde 1900 civarında CFTR mutasyonları olduğu bilinmektedir. Bu mutasyonlardan bazıları oldukça yaygın ve tedavilere cevap verebiliyorken bazıları ise nadir görülmektedir.
CFTR’ın işlevi nedir?
CFTR geninin kodladığı protein, bir hücredeki NaCl dengesini sağlar. Hücrenin içindeki iyonların hücre dışına çıkması ve tuz dengesi ile doğru orantılı olarak suyun varlığını kontrol eder. Hücrelerimizde hücre içi ve dışı arasındaki bağlantıyı sağlayan geçiş kapıları bulunur. Bu çeşitli kapılardan molekül geçişleri yaşanır. Bu geçişlerde klor iyonları da taşınır ve bu geçidin yapımından sorumlu olan proteini CFTR salgılar. Nükleustan alınan kodlara göre transkripsiyon yapılır ve endoplazmik retikulumda olgunlaşmamış geçitler olan protein sentezlenir. Sonrasında golgi cisimciğine uğrayan bu geçit proteinleri burada olgunlaşarak hücre zarına ilerler ve fonksiyonlarına uygun çalışmaya başlar.
içindeki iyonların hücre dışına çıkması ve tuz dengesi ile doğru orantılı olarak suyun varlığını kontrol eder. Hücrelerimizde hücre içi ve dışı arasındaki bağlantıyı sağlayan geçiş kapıları bulunur. Bu çeşitli kapılardan molekül geçişleri yaşanır. Bu geçişlerde klor iyonları da taşınır ve bu geçidin yapımından sorumlu olan proteini CFTR salgılar. Nükleustan alınan kodlara göre transkripsiyon yapılır ve endoplazmik retikulumda olgunlaşmamış geçitler olan protein sentezlenir. Sonrasında golgi cisimciğine uğrayan bu geçit proteinleri burada olgunlaşarak hücre zarına ilerler ve fonksiyonlarına uygun çalışmaya başlar.
CFTR geni, geçitlerin yapılması ve açılıp kapanmasını kontrol etmekle beraber geçitlerin genişliğini de belirler. Sağlıklı bir insanda bu geçitler sayesinde hücrenin içinden dışına klor geçişi olur. Klor geçişi ile birlikte hücre dışının konsantrasyonu artar ve bu artış su moleküllerini çeker. Bu mekanizma sayesinde mukus sağlayan her organ ve sistemde mukusların akışkanlık ve yapışma oranı ayarlanır. Klor iyonlarının hücre dışına taşınması su moleküllerini çektiği için mukuslar daha sulu hale gelip kayganlaşır ve amaçlarına uygun hale gelirler.
CFTR Mutasyonunun Sonuçları
CFTR geninde mutasyon olan bir kişi ise mutasyon türüne bağlı olmamakla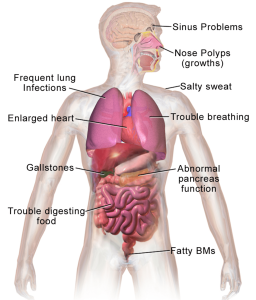 birlikte su-tuz dengesini sağlayamaz. Bu yüzden başta solunum sistemi, gastrointestinal, ürogenital sistem ve ekzokrin bezler olmak üzere vücuttaki hemen hemen her sistem etkilenir. Bu etki mukusun viskozitesinin artması sonucu salgılanan kanalları tıkamasıdır. Akciğer, pankreas, bağırsak ve karaciğerde sık görülme nedeni ise bol mukoza salgısı olan organlar olmalarıdır. Bu mukusun birikmesiyle bakteriler gibi patojen mikroorganizmaların üremesi ve enfeksiyon oluşturması oldukça kolaylaşır.
birlikte su-tuz dengesini sağlayamaz. Bu yüzden başta solunum sistemi, gastrointestinal, ürogenital sistem ve ekzokrin bezler olmak üzere vücuttaki hemen hemen her sistem etkilenir. Bu etki mukusun viskozitesinin artması sonucu salgılanan kanalları tıkamasıdır. Akciğer, pankreas, bağırsak ve karaciğerde sık görülme nedeni ise bol mukoza salgısı olan organlar olmalarıdır. Bu mukusun birikmesiyle bakteriler gibi patojen mikroorganizmaların üremesi ve enfeksiyon oluşturması oldukça kolaylaşır.
Bu yüzden kistik fibroziste ciddi akciğer enfeksiyonları sık görülür, hatta bazı hastalarda hastalığa işaret eden ilk semptomdur. Akciğer enfeksiyonlarından doğan komplikasyonlar ise genelde ölüme yol açan başlıca nedendir.
Mutasyona uğramış genin ifade ettiği proteinin hücreye 3 şekilde etkisi olabilir; hücre içi ve dışını bağlayan geçitlerin olmaması, bu geçitlerin düzgünce açılıp kapanmaması ya da iyonların geçemeyeceği kadar dar açılması. 1900’den fazla mutasyonda bu üç işlevsizlikten biri görülür. En sık karşılaşılan ve bu yüzden tedavisinde en fazla yol kat edilen mutasyon çeşidi ise ∆F508 mutasyonudur. Bu mutasyonda üç adet bazın silinmesi sonucu 508. sıradaki fenilalanin aminoasidi eksik kodlanmış olur. ∆F508 mutasyonunun yanı sıra ∆IF507 ve F508C ülkemizde karşılaşılan mutasyonlardandır.
CF Tanı ve Tedavisi
CF her ne kadar tehlikeli bir hastalık olsa da, erken tanı ve kişiye özel tedavi prosedürleri ciddi farklar yaratmaktadır. Hastalığın hayati tehlikeye yol açan en büyük sebebi olan enfeksiyonların oluşmadan veya yayılmadan önleminin alınması hastalığın gidişatını değiştirebilir. Bu yüzden erken tanı yapılması oldukça önemli ve kritiktir.
CF testinin komplike olabileceği düşünülebilir, fakat hastalığın teşhisinde başvurulan ilk test ter testidir. Hastadan alınan ter numunesinin içerdiği tuz miktarı beklenen referans değerlerinin üstündeyse tanı koyulmaktadır. Üstelik hızlı ve acısız bir yöntemdir. Bu işlem sonucunda teşhis konulup ileri tetkiklerle hastalığa neden olan mutasyonları anlayabilmek için gen inceleme testlerine başvurulabilmektedir. Genetik mutasyonlara göre tedaviye yanıt verme oranı değişmektedir.
Tedavide Güncel İlaç Kullanımı
Günümüzde CF için FDA tarafından onay almış ilaçların sayısının fazla olmadığını söylemek mümkün. Farklı mutasyonların her ilaca yanıt vermemesi söz konusu olduğu için piyasadaki ilaçların çoğu yaygın görülen mutasyonlara etki eden ilaçlar. FDA onayı almış ilaçlar ve çalışma mekanizmaları ise şu şekildedir:
• CAYSTON (aztreonam)
Cayston, inhalasyon suretiyle alınmak için hazırlanmış solüsyon halinde bir ilaçtır. İlacın içerdiği etken madde aztreonam bir antibakteriyaldir. Aztreonam, in vitro koşullarda gram-negatif aerobik patojenlere etki gösterir. CF’te karşılaşılan enfeksiyonlara yol açan patojenlerden en sık karşılaşılanlarından olan P. Aeruginosa adlı bakteride etki gösterir. Aztreonam antibiyotik varlığında üremesine devam edemeyen bakterilerin, penicilline bağlanan proteinlerine bağlanır. Bu bağ hücre duvarı sentezinin inhibisyonu ve hücre ölümü ile sonuçlanır. Cayston CF mekanizmasına etki etmez, solunum yollarındaki enfeksiyonların neden olduğu semptomları iyileştirmek için kullanılır.
ilaçtır. İlacın içerdiği etken madde aztreonam bir antibakteriyaldir. Aztreonam, in vitro koşullarda gram-negatif aerobik patojenlere etki gösterir. CF’te karşılaşılan enfeksiyonlara yol açan patojenlerden en sık karşılaşılanlarından olan P. Aeruginosa adlı bakteride etki gösterir. Aztreonam antibiyotik varlığında üremesine devam edemeyen bakterilerin, penicilline bağlanan proteinlerine bağlanır. Bu bağ hücre duvarı sentezinin inhibisyonu ve hücre ölümü ile sonuçlanır. Cayston CF mekanizmasına etki etmez, solunum yollarındaki enfeksiyonların neden olduğu semptomları iyileştirmek için kullanılır.
• PULMOZYME (Dornase alfa)
Pulmozyme de cayston gibi solunum yollarındaki enfeksiyonlara ilişkin kullanılır. Farklı olarak Pulmozyme, kıvamı kalınlaşmış olan mukozayla başa çıkmak için kullanılır. Bu ilaç inhalasyon suretiyle alınmak üzere hazırlanmış solüsyon olarak tek kullanımlık ampullerde bir nebülizatör aracılığıyla vücuda alınır. İçindeki etken madde dornase alfa rekombinant insan deoksiribonükleaz enzimidir. Bu enzimin klinik öncesi in vitro çalışmalarda DNA’nın bazı yerlerini parçaladığı görülmüştür. Pulmozym, hastalarda biriken mukustaki DNA’ları parçalayarak mukusun viskozitesini azaltır..
• TOBİ (Tobramycin)
Tobi de bahsettiğimiz iki ilaç gibi inhalasyon suretiyle kullanılan bir antibiyotiktir. Diğerlerinden farklı olarak 1975’ten beri intravenöz formu da bulunmaktadır. CF’te en fazla görülen enfeksiyonlardan biri olan P. Aeruginose tedavisinde kullanılır.
• ZENPEP (Pancrelipase)
Farklı adlarla bulunabilen bu ilacın etken maddesi olan pancrelipase enzimi, doğal olarak pankreastan duodenuma salgılanan, karbonhidrat, yağ ve protein gibi besin maddelerinin parçalanması sağlar. Bu enzim pankreastan ince bağırsağa pankreatik (Wirsung) kanalı ile iletilir. Bu iletim esnasında CF ile ilişkili olan herhangi tıkanıklık sonucu yeterli miktarda enzim duodenuma ulaşamaz ve besin malabsorpsiyonu oluşur. Sadece CF’te değil aynı zamanda pankreatit gibi sindirim sisteminde etkili organların tahribatında da kullanılan bir ilaçtır. Pankreasın doğal çalışmasını mimikler. Alınan kapsüller midede açılır ve ince bağırsağa kadar enterik kaplamaları sayesinde çözünmez. Duodenuma ulaştığında pH yükseldiği için çözünür ve etki göstermeye başlar. İlaç kesinlikle yemeklerle beraber kullanılmalı ve çiğnenip ezilmemelidir.
• KALYDECO(Ivocaftor)
 Ivocaftor kendi başına bir ilaç olarak kullanılmakla beraber çeşitli etken madde kombinasyonlarıyla birlikte farklı ilaç formülasyonlarında da görülmektedir. Bu ilacın çalışma mekanizmalarını anlamak için biraz daha teknik detaylara girmek gerekiyor.
Ivocaftor kendi başına bir ilaç olarak kullanılmakla beraber çeşitli etken madde kombinasyonlarıyla birlikte farklı ilaç formülasyonlarında da görülmektedir. Bu ilacın çalışma mekanizmalarını anlamak için biraz daha teknik detaylara girmek gerekiyor.
CFTR proteinin beş farklı yapısal mutasyonları bulunmaktadır. Class I mutasyonlarında herhangi bir fonksiyonel CFTR proteini üretilmemektedir. Class II mutasyonlarında ise protein üretilmektedir fakat girmesi gereken şekilden farklı olarak katlanır, bu yüzden hücrede parçalanır ve sentezlenenden daha az protein hücre yüzeyine ulaşır. Class III mutasyonlarında protein sentezlenip hücre zarına ulaşmakta fakat geçidin açılıp kapanma mekanizması regüle edilememektedir. Class IV mutasyonlarında da III gibi hücre zarına ulaşmakta fakat geçit fonksiyonları sıkıntı çıkarmaktadır. Class V mutasyonlarında protein sentezlenip hücre yüzeyine ulaşmakta fakat yeterli miktarda sentezlenmemektedir.
Bu 5 farklı tipten sadece Class III Ivocaftor’e olumlu cevap vermektedir. Diğer farklı mutasyonlar ise ya tedaviye yanıt vermiyor ya da başka moleküllerle kombinasyonu gerekiyor. Class II olan F508del mutasyonuna Ivocaftor tek başına etki etmemektedir. Ivocaftor’un çalışma mekanizması, iyon geçitlerinin daha sık açılıp kapanması ve daha uzun süre açık durması, bu sayede klor iyonlarının hücre dışına çıkışının artması yönündedir.
Yapılan klinik araştırmalarda Ivocaftor’un yağ açısından zengin besinlerle birlikte alınmasının tedavide fark edilebilir bir değişikliğe yol açtığı görüldü. Dozaj formu hastanın yaşı, eşlik eden diğer hastalıkları ve ciddiyetine göre değişmekte fakat genel olarak reçetelenen doz 12 saatte 1 olacak şekildedir. İlacın 4 aydan küçük bebeklerde etkisi ise bilinmemektedir.
• ORKAMBI(Luvacaftor-Ivocftor)
Bu ilaç, Ivocaftor’un tek başına yeterli olmadığı ve CF vakalarında en sık karşılaşılan mutasyonlardan biri olan F508del mutasyonu için kullanılmaktadır. 1 yaşından büyük ve F508del mutasyonunu homozigot olarak bulunduran hastalarda kullanılmaktadır. F508del mutasyonu Class II mutasyonlarına girmektedir. Lumacaftor CFTR proteinlerinin daha konfirmasyonel stabilitesini artırır, dolaylı olarak katlanmasını düzeltir ve hücre yüzeyine ulaşımını artırır.
• SYMDEKO(Tezacaftor-Ivocftor)
Bu kombinasyonda ise tezacaftor, daha fazla CFTR proteinlerinin yüzeye ulaşmasını sağlarken, ivocaftor ise geçitlerin daha uzun süre açık kalmasını sağlamaktadır. Orkambi’de olduğu gibi F508del mutasyonunu homozigot içeren bireylerde etkili olduğu bilinmektedir. 6 yaşında ve daha büyük bireylerin tedavisinde kullanılmaktadır.
ulaşmasını sağlarken, ivocaftor ise geçitlerin daha uzun süre açık kalmasını sağlamaktadır. Orkambi’de olduğu gibi F508del mutasyonunu homozigot içeren bireylerde etkili olduğu bilinmektedir. 6 yaşında ve daha büyük bireylerin tedavisinde kullanılmaktadır.
• TRIKAFTA(elexacaftor-tezacaftor-ivocaftor ve ivocaftor)
Trikafta en 1 adet F508del aleli bulunduran hastaların tedavisinde kullanılmaktadır. Trifakta’nın içerdiği elexacaftor tezcaftor benzer bir çalışma mekanizmasına sahip. Fakat tezacaftorden farklı bağlanma bölgeleri kullanarak CFTR proteinin hücre yüzeyindeki işlevselliğini arttırmaktadır. Bu üç etken madde bir arada kullanıldığında F508del başta olmak üzere geniş bir spektrumdaki mutasyonlarda etkisi artmış olmaktadır. Trikafta da Symdeko gibi 6 yaşından büyük hastaların tedavisinde reçete edilmektedir.
KAYNAK
* Ivacaftor: A Novel Gene-Based Therapeutic Approach for Cystic Fibrosis Michelle E. Condren, PharmD1,2 and Marquita D. Bradshaw,
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3626070/
*Ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation Isabelle Sermet-Gaudelus European Respiratory Review Mar 2013,
https://err.ersjournals.com/content/22/127/66
* Rafeeq, M.M., Murad, H.A.S. Cystic fibrosis: current therapeutic targets and future approaches. J Transl Med 15, 84 (2017).
https://doi.org/10.1186/s12967-017-1193-9
https://www.cff.org/research-clinical-trials/basics-cftr-protein
https://www.cff.org/research-clinical-trials/types-cftr-mutations
https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-017-1193-9
https://www.centerwatch.com/directories/1067-fda-approved-drugs/topic/175-cystic-fibrosis
https://www.cff.org/node/1316
https://www.cff.org/sites/default/files/2021-12/Know-Your-CFTR-Mutations-Infographic.pdf